
Recent Progress in Small-Molecule Tumor Necrosis Factor Receptors (TNFR) Modulators
Junyong Wang1#, Ruizhi Guo2#, Raorao Zheng1, Hongliang Li1, Dingru Qiao1, Jiansong Wang2,3*, Yingxia Bao2* and Jiaji Zhao1*
1School of Chemistry and Chemical Engineering, Guangdong Pharmaceutical University
2Guangzhou Baiyunshan Pharmaceutical Holdings Co., Ltd. Baiyunshan Pharmaceutical General Factory
3Guangzhou Caizhilin Pharmaceutical Co., Ltd.
Citation: Wang J, Guo R, Zheng R, Li H, Qiao D, et al. Recent Progress in Small-Molecule Tumor Necrosis Factor Receptors (TNFR) Modulators. Asian Journal of Complementary and Alternative Medicine, Vol 13(2), 30-43:2025.
Abstract
Tumor necrosis factor receptors (TNFR-) are emerging as an important class of pharmaceutical targets that have potential value for the treatment of numerous diseases, including cancer, inflammatory diseases, as well as autoimmune diseases. Although a lot of antibody reagents targeting TNFRs have entered clinical investigation, few of them have been approved for clinical use. More importantly, small-molecule TNFR- inhibitors have rarely entered clinical trials. This brief review focuses on the development of small molecular TNFR- modulators that have been reported in recent decades, with the aim of shedding light on the future discovery and application of more potent and promising small-molecule TNFR modulators.
Tumor necrosis factor receptors (TNFR) are receptors for tumor necrosis factor α (TNF-α). There are two types of TNFR: TNFR1 and TNFR2 [1]. Both TNFR1 and TNFR2 are type I transmembrane proteins, which have an N-terminal extracellular domain (ECD) with four cysteine-rich domains (CRDs), a transmembrane domain, and a cytoplasmic domain. TNFR1 is expressed ubiquitously in various cell types [2], while TNFR2 is expressed mainly in tumor cells and immunosuppressive cells, such as regulatory T cells (Tregs) [3-5], mesenchymal stem cells (MSCs) [6], oligodendrocytes [7], and myeloid-derived suppressor cells (MDSCs) [8].
There are some differences between TNFR1 and TNFR2. The most distinctive difference is that the intracellular part of TNFR1 contains a death domain (DD), which can recruit TNF receptor-associated death domain (TRADD) and receptor-interacting serine/threonine protein kinase 1 (RIPK1). On the other hand, TNFR2 has no death domain. Another difference is that the main ligand of TNFR1 is soluble TNF (sTNF) [9], while that of TNFR2 is membrane TNF (mTNF) [10].
TNFRs are involved in signaling pathways of numerous chronic diseases (Scheme 1) [11]. The recruitment of TRADD by TNF-α/TNFR1 complex will lead to activation of nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) in a canonical pathway [12], and subsequently activate the anti-apoptotic genes such as c-FLIP. Abnormal activation of TNFR1 often lead to autoimmune responses [13], chronic inflammation such as rheumatoid arthritis (RA) [14] and inflammatory bowel disease (IBD) [15], infectious diseases [16], and cancer [17]. On the other hand, the combination of TNF and TNFR2 causes recruitment of TNF receptor-associated factor 1/2/3 (TRAF1/2/3) and cellular inhibitor of apoptosis (cIAPs), and activate non-canonical NF-κB pathway [18-19]. These will initiate signaling pathways that lead to the survival and proliferation of regulatory T cells (Tregs), which is in charge of immune suppression. In recent years, TNFR2 has emerged as a promising target in two major classes of disease that threaten public health: inflammatory diseases [9] and tumor immunotherapy [20-26].
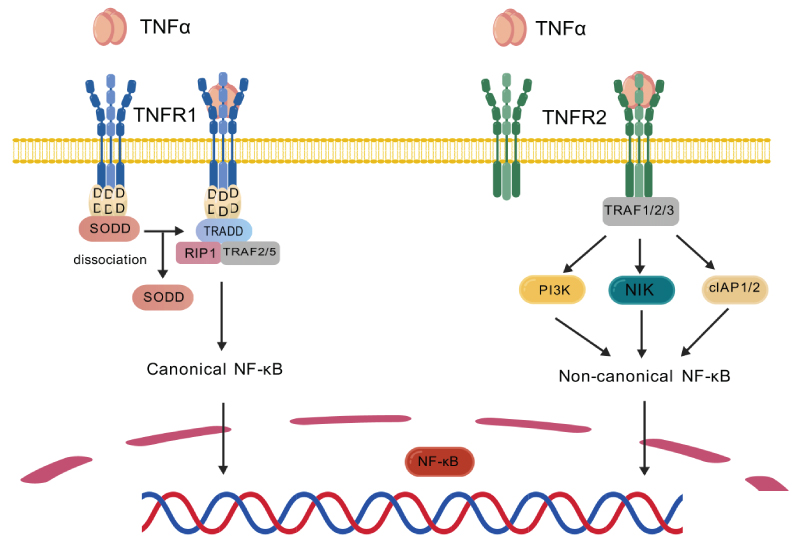
Scheme 1: Singal Pathways involved in TNFRs.
2. Advantages and Challenges of Small-Molecule TNFR Modulators
Given the important role of TNFRs in the process of immunosuppression, much effort has been devoted to the development of TNFR mediators. However, to date, no TNFR mediators have been approved for clinical use. Most candidates that are undergoing clinical investigations are antibodies, whereas small-molecule TNFRs mediators have rarely progressed to clinical trials. Although antibody drugs are highly specific for extracellular targets, they also have some limitations: the need for invasive administration; poor membrane permeability; limited tissue penetration; low stability and high production/transportation costs. Compared with antibodies, small-molecule TNFR modulators offer several advantages: oral administration (improving patient compliance), stronger membrane permeability, higher stability, and lower production/transportation costs. However, some challenges have been encountered in the development of small-molecule TNFR modulators:
(1) The binding interface between TNFRs and their ligands (e.g. TNF-α) is relatively large and lacks deep binding pocket, making it challenging for small molecules to achieve efficient competitive inhibition. Consequently, the design of traditional inhibitors faces significant difficulties.
(2) TNFRs are transmembrane proteins, requiring small molecules to simultaneously meet both domain binding and membrane localization requirements, thus presenting high design challenges.
(3) Cross-reactivity between TNFR1 and TNFR2 may lead to severe toxicity (e.g. systemic inflammation), as TNFR1 mediates pro-inflammatory and apoptotic signaling, whereas TNFR2 is primarily involved in proliferation and immune regulation.
Despite these challenges, some small-molecule TNFR modulators have been reported to show promising in vitro and in vivo bioactivity [27-28]. In this brief review, we will focus on small-molecule TNFR agonists and antagonists reported in recent years.
3. Key Strategies in Small-Molecule TNFR Modulators Discovery
Several strategies have been successfully applied to the development of TNFR modulators, including disrupting TNF-α trimerization, screening for allosteric modulators (e.g., molecules that stabilize inactive conformations) of TNFRs, and binding pocket analyses of TNFRs with the aid of advanced AI technologies.
(1) Disrupting TNF trimerization: since trimerization is essential for the binding of TNF-α to TNFRs, small molecules that disrupt the TNF trimer would block the TNF-TNFR pathway. [29] Although this strategy did not directly target TNFRs, it proved effective for treating TNFR-induced inflammatory diseases.
(2) Allosteric TNFR modulators: allosteric modulators may hinder the assembly of TNFR1 through binding to the pre-ligand-binding assembly domain (PLAD), preventing it from forming dimers which accept TNF-α trimer. As PLAD is an extracellular subunit of TNFRs, ligands need not penetrate the cell membrane to exert their effects.
(3) AI-assisted pocket analysis of TNFRs: with the aid of AI tools such as AlphaFold [30] and FTMap [31], the full-length 3D structure of unliganded TNFRs could be generated, and the dynamic Pre-ligand-binding assembly domain (PLAD)-PLAD interaction was revealed. Consequently, most likely ligand-binding pockets were identified, facilitating the design of modulators.
4. Modulators of TNFR1 Signal Pathway
As early as 1997, Murali et al. developed several exocyclic peptidomimetics that inhibit TNF-α binding to TNFR1 using structure-based design [32]. In this pioneering study, the author identified three binding sites of TNFR1 based on crystal structures of TNF-β/TNFR1 complex [33] and TNFα: loop 1 of domain 2 (Phe60-His66), loop 2 of domain 2 (Cys76-Val83), and loop 1 of domain 3 (Trp107-Leu111). The critical peptide fragments of these binding sites were used as templates for the design of exocyclic peptidomimetics as TNF-α antagonist candidates. The most potent peptidomimetics, WP9QY, which mimics loop1/domain3 of TNFR1, exhibited an IC50 of 5μM against TNF-α. In vivo study proved that WP9QY inhibited collagen-induced increases in the arthritis score, but its anti-inflammatory effect was weaker than that of anti-TNF antibody [34]. In 2014, Ma et al. used a similar strategy to report a novel class of small-molecule TNF-α inhibitors that block the TNF/TNFR1 interaction. Among these compounds, the lead compound, C87, inhibited TNF-α induced cytotoxicity (IC50=8.73 μM), attenuated TNF-α induced inflammation and liver injury in animal models [35]. The discovery of C87 was facilitated by computer aided virtual screening of 90000 compounds, for molecules that mimic the peptide fragments on the TNFR1 loop2/domain2 (amino acid 77-83), which was identified as the key region for TNF/TNFR1 interaction [32-33]. Structure-activity relationship (SAR) indicated that the C-N double bond with E-configuration, as well as the 3-nitro-4-chloro phenyl group were essential for activity. However, C87 failed to effectively block TNF-α binding to TNFR1/2 in ELISA. In the hepatitis mouse model, survival rate of the C87 treated group was lower than that of Enbrel. Thus, directly mimicking TNFR1 peptide fragments might not be the most efficient strategy for TNF/TNFR1 signaling blockade.
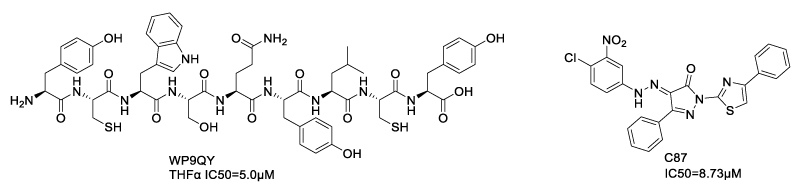
Scheme 2: TNF-α inhibitors based on mimicking peptide fragments of TNFR1 binding sites.
Since the TNF-α trimerization is essential for its binding to TNFRs, an efficient strategy to block TNF-α/TNFR transduction was to develop TNF-α ligands capable of disrupting its trimerization (Scheme 3). In 2005, He et al discovered a small-molecule TNF-α inhibitor named SPD304, which strongly inhibited TNF/TNFR1 interaction and the downstream IκBα degradation in HeLa cells [36] (Shceme 3). SPD304 featured two aromatic hydrophobic fragments linked by a dimethylamine spacer. X-ray crystallography of TNF-α with SPD304 revealed that SPD304 completely replaced a subunit from TNF-α trimer which is necessary for TNFR1 binding, resulting in a TNF-α dimer-SPD304 complex. This dimer was composed of 16 contact residues, including 9 residues from chain A and 7 residues from chain B. The trifluoromethylphenyl indole and the dimethyl chromone moieties of SPD304 folded back upon one another. This conformation enabled SPD304 to fit into the TNF-α subunits’ interface, providing insights into rational molecule design. Using a fluorescence homoquenching assay, the authors demonstrated the mechanism of TNFα-SPD304 formation was binding of SPD304 with the TNFα trimer at the first step and then accelerating the dessociation of a subunit.
Using a similar strategy, Lai et al. also discovered a series of TNF-α inhibitors by TNF-α dimer-based virtual screening [37]. The most potent compound, AP-906/41640035 (Scheme 3), exhibited an IC50 of 14.0±0.6 μM. The blockade of TNF-α/TNFR1 interaction by AP-906/41640035 was confirmed by surface plasmon resonance (SPR)-based in vitro competitive binding assay. SAR study disclosed that the naphthothiophen-2-one moiety formed hydrophobic interaction with Y59B, L57A and L57B, while the two phenyl groups formed π-π stacking with Y119A,Y119B and Y151B. The sulfonyl group formed hydrogen bonds with polar residues Q61B. The reason that AP-906/41640035 was superior to other compounds was that it can form multiple π-π stacking networks with the binding site. Recently, Lin and Li et al. disclosed a novel class of pyrrolidine-2,5-dione TNF-α inhibitors, which bind to the TNF-α dimer and block TNF-α/TNFR triggered NF-κB signaling pathway [38]. The best candidates, compound 0 and compound 1 (Scheme 3), were characterized by two flexible aromatic side chains.
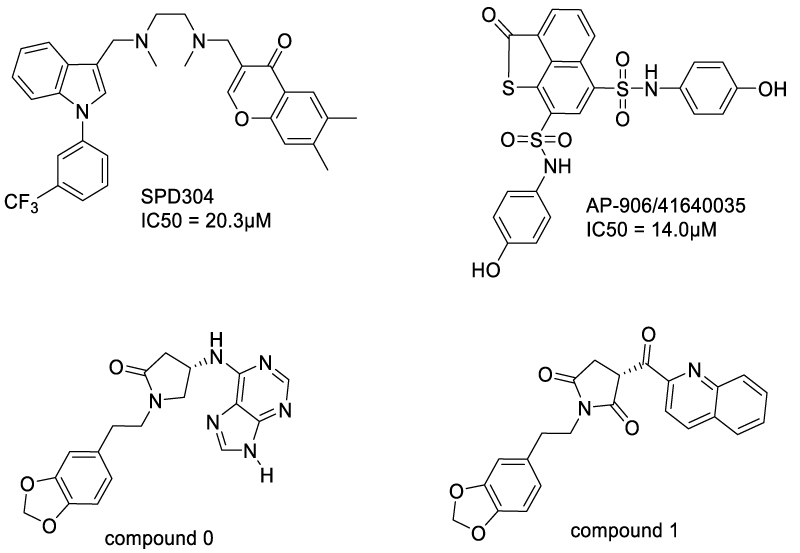
Scheme 3: Molecules bind to TNF-α dimer and accelerate subunit dissociation of TNF-α trimer.
Another important strategy for designing TNF-α ligands that inhibit TNF-TNFR interaction is to distort the TNF-α trimer, so that TNFR binding was disturbed. In 2019, O’Connell et al. reported a series of small molecules capable of impairing the TNF-TNFR1 signaling pathway, by stabilizing the asymmetric form of TNF-α trimer [39] (Scheme 4). Among these compounds, UCB-6876 exhibited a KD of 22 μM with TNF. Crystal structure of UCB-6876/TNFR1 complex revealed that three TNFR monomers formed an asymmetric trimeric unit, wherein the UCB-6876 bound at the center of the TNF trimer. This binding stabilized the asymmetric form of TNF-trimer, thus inhibiting the TNFR1 signaling and down-regulating downstream proteins such as RIP-1 and pNF-κB. The key interactions of UCB-6876 with TNF trimer were hydrogen bond between benzimidazole N atom and Y151C, and π stacking of benzimidazole with Y59C. The 2,5-dimethylbenzyl moiety of UCB-6876 inserted into a hydrophobic pocket formed by Y59A, Y119B and L57B. By modifying UCB-6876’s hydroxymethyl group with a pyridyl moiety, an additional hydrogen bond was formed with Y119A, significantly enhancing its potency (KD = 9 nM). Adding pyrazole substituent on the benzimidazole ring gave UCB-9260, which bound to TNF with similar affinity (KD = 13 nM), but exhibited a slower dissociation rate. In 2021, O'Connell further optimized molecules that distort TNF trimer and reduce the binding affinity of TNF with the third TNFR1 molecule, and discovered UCB-4433 and UCB-0595 [40] (Scheme 4). Furthermore, the crystal structure of TNF-TNFR1 complex with UCB-4433 was obtained, revealing distortion at the A-C receptor binding site.
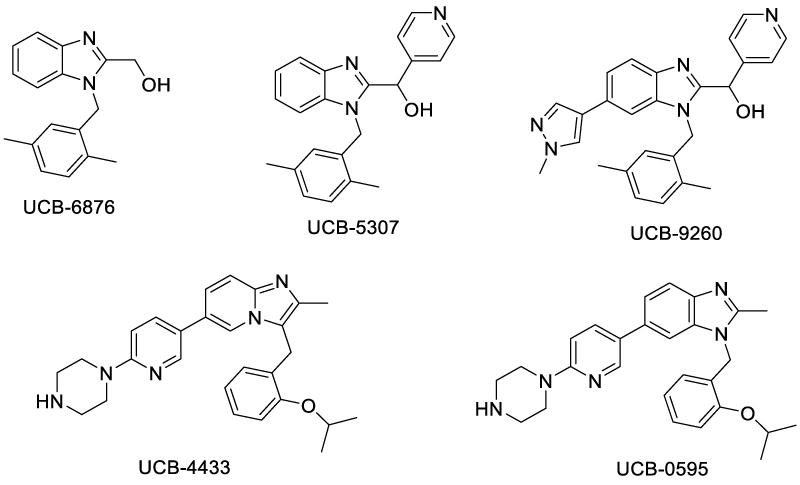
Scheme 4: Small molecules that distort the TNF-α trimer.
After extensive optimization, O'Connell et al. reported a novel TNF inhibitor that blocks TNFR1 signaling pathway, SAR441566, for the treatment of rheumatoid arthritis (Scheme 5). SAR441566 exhibited a KD of 15.1 nM, with improved drug-like properties according to Lipinski’s rule of five [41]. This molecule was later named balintumfib for phase I clinical trials.
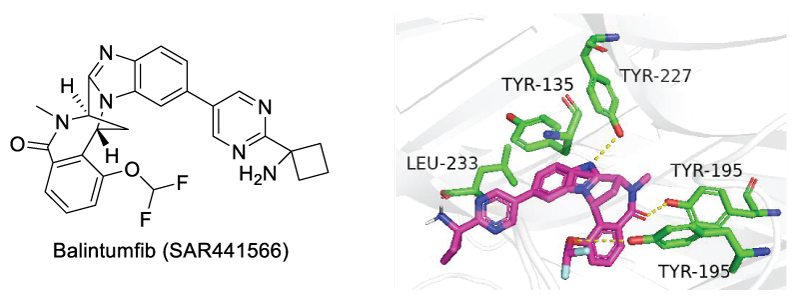
Scheme 5: Chemical Structure of Balintumfib (left) and its binding site in sTHF-α (right).
Balintumfib (SAR441566) is the first oral small-molecule TNF-α/TNFR1 signaling inhibitor that has entered clinic trials. Mechanistically, Balintumfib stabilizes an asymmetrical form of soluble TNF-α, preventing the higher-order receptor clustering required for TNF-mediated signaling. Compared with other TNF-α ligands, Balintumfib features the rigid bicyclic structure, which contributes to its drug-like properties [42], including strong tissue penetration, acceptable oral bioavailability, and favorable distribution. Docking model of Balintumfib with TNF-α indicated π-stacking interaction with Tyr135, and several hydrogen bonds with Tyr227 and Tyr195 (Scheme 5, right) [43]. In the ascending dose study in healthy male participants, single (5-600 mg) and multiple (100-600 mg) oral dose of Balintumfib were well tolerated in all participants [44]. Pharmacokinetics (PK) analysis showed a median tmax of 2.5-5 hours, mean terminal half-life of 22-30 hours, and a time to reach steady state of 5-6 days. Pharmacodynamic (PD) assessment showed complete TNF-α receptor occupancy at all tested time points. These results indicated a favorable safety profile and PK/PD characteristics of Balintumfib.
A convenient way to develop TNFR inhibitors was starting from approved drugs. In 2017, Lo et al. identified two drugs with TNFR1 inhibitory activity, zafirlukast and triclabendazole [46], using a novel high-throughput fluorescence lifetime screening system (Scheme 7). After HTS over 446 compounds from NIH clinical collection, the two drugs were found to decrease FRET efficiency in a dose-dependent manner. The IC50 of zafirlukast and triclabendazole were 18 μM and 15 μM, respectively. In 2022, Lin et al. reported atrazine (ATR) as a ROS/TNF-α/TNFR1 pathway activator [47] (Scheme 7). By inducing oxidative stress in L8824 cells, ATR promoted the binding of TNF-α and TNFR1, activating apoptosis and necrosis through the TNF-α/TNFR1 pathway. The effect of atrazine upon TNF-α/TNFR1 could be antagonized by tannic acid.
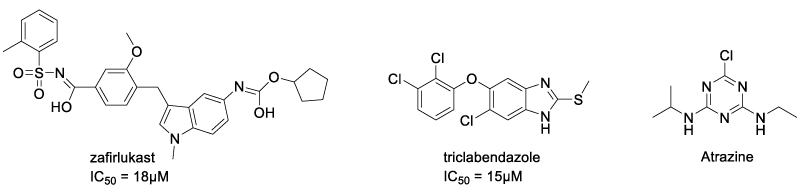
Scheme 8: Chemical Structure of zafirlukast and R1.
Allosteric TNFR1 ligands can efficiently disturb the binding interface of TNFR1 with TNF. In 2005, Murali et al. reported a TNFR1 modulator, F002, which disabled TNFR1 function by inducing allosteric modulation of tryptophan-107 in TNFR1 [49] (Scheme 9). F002 reduced TNF-α induced cytolysis of murine L929 and attenuated TNF-α induced downstream signaling, such as phosphorylation of IKBα and P38. It was found that F002 bound selectively to the allosteric pocket through hydrophobic interaction, with high affinity (KD = 0.45 μM). Residues 82Q and 112F were identified to be key residues contributing to this interaction. This binding induced a perturbation of the WP9 loop, which is the key region for TNF-α/TNF interaction. SAR investigation of F002 proposed the necessity for rigid propeller-like arrangement of aryl groups. To improve the solubility, Rowe et al. combined the propeller-like aryl groups of F002 with propane diol backbone to identify a novel TNFR1 inhibitor C7 with higher docking score than F002. To lower the molecular weight and LogP, the benzyl alcohol acetates of C7 were removed, yielding SGT11, which had higher docking score and better lipophilicity [50]. As a potential therapeutic candidate for traumatic brain injury, SGT11 occupied TNFR1 cavity at the TNF-binding interface. In vitro evaluation indicated that SGT11 inhibited IκB phosphorylation. SGT11 also demonstrated in vivo efficacy, mitigating post-traumatic sleep disturbances and improving functional recovery.
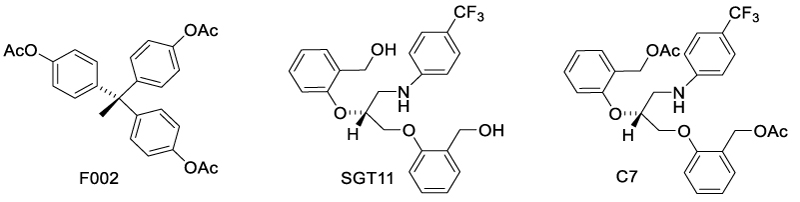
Scheme 9: Allosteric TNFR1 modulators that disturb the binding interface.
In 2019, Lo et al. reported several TNFR1 allosteric modulators, including DS41, DS114, and SB-200646 (Scheme 10). While DS41 and DS114 were allosteric inhibitors [41], SB-200646 was an allosteric activator [51]. Unlike previously reported modulators, these modulators bound in the intermonomeric space between TNFR1 dimers, inducing conformational changes without affecting receptor-receptor or receptor-ligand interactions [52-54]. Impressively, these compounds showed promising drug-like properties and blood-brain-barrier penetration capabilities.
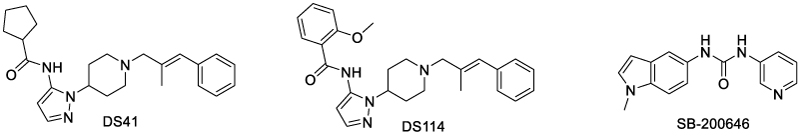
Scheme 10: Allosteric TNFR1 modulators binding in the interspace between TNFR1 dimers.
In 2019, Huang et al. identified several TNFR1 and TNFα-TNFR1 inhibitors using pharmacophore-based virtual screening [54]. By searching the ZINC purchasable compound database with TNFR1 and TNFα-TNFR1 query features, 37 and 45 hits were obtained respectively. After molecular docking and ADMET prediction, 4 lead compounds were identified for TNFR1 and 6 for TNF-α/TNFR1 complex. However, in vitro assays were not performed to prove the outcome of virtual screening.
5. Modulators of TNFR2 Signal Pathway
Immune evasion is a phenomenon whereby tumor cells avoid identification and attack by immune system, survive and proliferate in the human body. Although the past decades have seen great progress in tumor immunotherapy [55], immune evasion has emerged as a huge challenge for effective immunotherapy treatment.
In the tumor microenvironment, TNFR2-induced overactivation of Tregs is an important cause of immune evasion [56]. Tregs inhibit the anticancer effect of CD8+ T cells through secretion of cytokines like IL-10 and TGF-β, which upregulate immune checkpoint proteins on CD8+ T cells [57], and other signaling pathways. It has been reported that TNFR2 over-expression in regulatory T cells promotes the progression of various malignant processes, including pleural effusion [58], hepatocellular carcinoma prognosis [59], and melanoma [60].
Compared with TNFR1, reports of small-molecule TNFR2 modulators are very rare, since target-based design of TNFR2 modulators is more challenging. TNFR1 contains a canonical "death domain (DD)", which directly recruits downstream signaling molecules (e.g., TRADD, FADD, and caspases) to mediate apoptosis and inflammatory signaling. Its ligand-binding pocket is relatively well-defined, making it more amenable to small-molecule targeting. In contrast, TNFR2 lacks the death domain and primarily signals through TRAF1/2 to promote survival and immune regulation. Its binding to TNF (or membrane-bound TNF) relies more heavily on protein-protein interactions (PPIs), making it more challenging for small molecules to disrupt.
Furthermore, TNFR2 is a high-affinity receptor for membrane-bound TNF (m-TNF), but not soluble TNF (sol-TNF). So far, most reported TNF ligands mainly target sol-TNF, but lack selectivity for m-TNF, so the m-TNF/TNFR2 signal transduction is difficult disrupt.
As early as 2002, Marriott et al. reported the inhibitory effect of thalidomide on T-cell surface expression of TNFR2 and soluble TNFR2 (sTNFR2) levels [61]. However, thalidomide had no effect on total (surface/intracellular) TNFR2 protein expression, suggesting that it actually inhibits the TNFR2 trafficking to the cell membrane.
In 2010, the crystal structure of TNF/TNFR2 complex was first reported by Mukai et al. [62], opening the gate for rational design of inhibitors targeting TNF/TNFR2 signaling pathway (Scheme 11). The author found that in the TNF-TNFR2 complex, TNF formed a central homotrimer, around which three TNFR2 molecules were bound. Each TNFR2 molecule interacts with two TNF subunits. Although the interface between TNF and TNFR2 is large, two core regions were identified, namely region 3 and 4. Region 3 features a cluster of acidic residues including Asp54, Glu57, and Glu70 and a negatively charged surface, while region 4 features a cluster of basic residues including Arg77, Lys108, and Arg133 and a positively charged surface. These elucidations shed light on the rational design of small-molecule TNFR2 ligands.
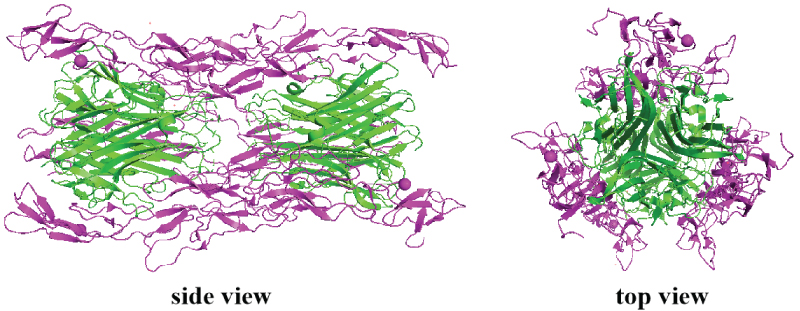
Scheme 11: Crystal Structure of TNF-TNFR2 complex (PDB ID: 3alq).
In a virtual screening of over 400000 natural compounds from the Traditional Chinese Medicine Database, Siu and Shaikh et al. identified 8 compounds with top quantitative protein-ligand interaction descriptor (QPLD) scores [63]. Among these ligands, five were region 3 (Scheme 12) binders and three were region 4 binders (Scheme 13). The authors proposed that region 3 was more favorable for TNFR2 inhibitors than region 4, since TNF binds much more tightly in region 4 than in region 3, so ligands in region 3 was more competitive. Molecular dynamics simulation showed that the top-scoring rigion 3 binders formed hydrogen bonds with Asp54, Cys 71 and Glu 57, which are key residues in the TNF-TNFR2 interaction. However, the authors did not perform experimental assay for the top hits.
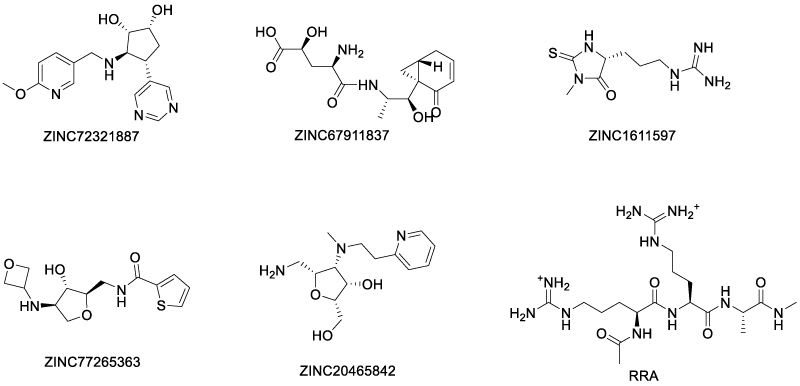
Scheme 12: Top score region 3 binders of TNFR2 by virtual screening (ref. 54).
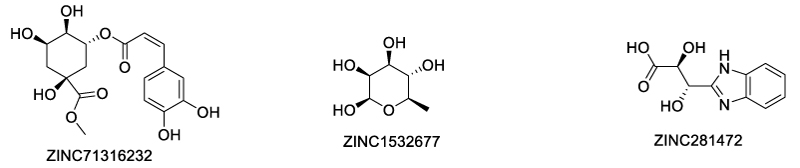
Scheme 13: Top score region 4 binders of TNFR2 by virtual screening (ref. 54).
In 2020, Chen et al. demonstrated that some Ca2+ channel inhibitors can promote the expansion of TNFR2-expressing CD4+Foxp3+ regulatory T cells (Tregs) and inhibit autoimmune inflammatory responses [64] (Scheme 14). Among them, a component from Chinese herbal medicine extracts, tetrandrine, which was an L-type calcium channel [65] and two-pore channel (TPC) [66] inhibitor, promoted the proliferation of Tregs most significantly. As for the mechanism, m-TNF can be cleaved to sol-TNF by TNF-converting enzyme (TACE), and tetrandrine down-regulates membrane-bound TACE expression on dendritic cells through inhibition of TPC, consequently reduced the expression of in-solution TNF (sol-TNF) while increasing the expression of m-TNF. As m-TNF preferentially binds to and activates TNFR2 which is expressed by Tregs, the m-TNF/TNFR2 interaction and subsequent expansion of Tregs were promoted. In vivo evaluation showed tetrandrine was promising for treating inflammatory diseases and autoimmune diseases. Besides tetrandrine, three other L-type channel inhibitors, namely diltiazem, nimodipine, and verapamil, have similar effects on the expression of m-TNF by dendritic cells (DCs). Since all of these compounds inhibit calcium signaling triggered by nicotinic acid adenine dinucleotide phosphate (NAADP), the authors proposed that NAADP-mediated signaling might connect with m-TNF expression, and Treg proliferation. This was demonstrated by the capacity of NAADP specific inhibitor Ned 19 to up-regulate TNF and down-regulate sol-TNF expression by bone marrow-derived DCs. As TPCs are the major calcium channel activated by NAADP, the effect of NAADP inhibitors on tmTNF expression was most probably through TPCs, as demonstrated by the fact that TPC siRNA also resulted in the upregulation of mTNF expression by DCs.
In 2022, Chen et al. disclosed that another natural product from Chinese herbal medicine, scutellarin, reduced TNFR2-expressing CD4+ Foxp3+ regulatory T cells (Tregs), enhancing anti-tumor immune responses [67]. It was found that scutellarin disrupted the interaction between TNF-α and TNFR2, inhibiting the downstream signaling pathway, such as p38 MAPK phosphorylation. In vivo assessment also indicated that scutellarin enhanced the effects of immunotherapy in a mouse CT26 colon cancer model. The inhibition of scutellarin on TNFR2 was further demonstrated by Mei et al in 2023, where scutellarin reduced triple-negative breast cancer (TNBC) metastasis by alleviating the TNF-α induced G-CSF expression. Inhibition of TNF-α / TNFR2 interaction was achieved by binding of scutellarin to TNFR2 in Endothelial cells [68]. Molecular docking showed scutellarin formed hydrogen bonds with Ser65, Ser76, Arg77, Ser79, and Asp140 residues in the region 4 of TNFR2, preventing TNF from binding to TNFR2 (Scheme 15). Scutellarin also inhibited the nuclear translocation of runt-related transcription factor 1 (RUNX1), which was found to bind to the promoter of G-CSF in TNBC tumor vessels and regulated G-CSF expression.
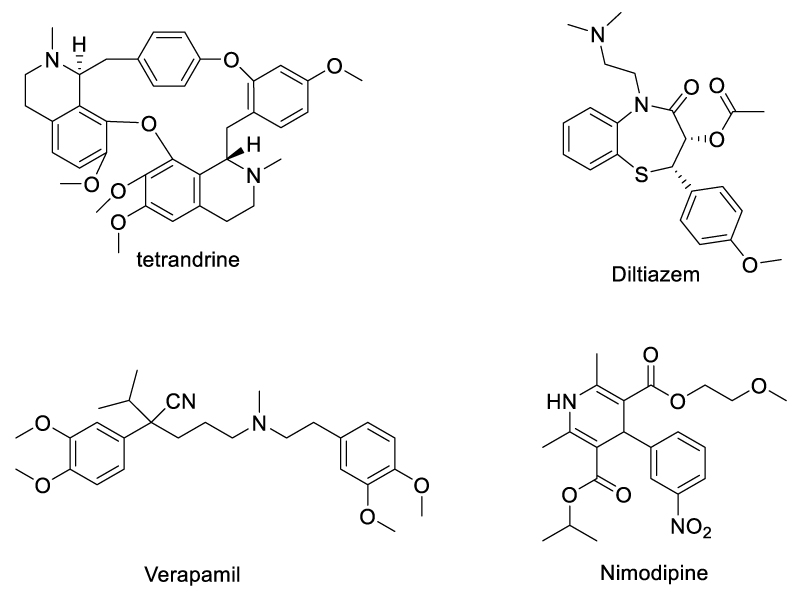
Scheme 14: TPC Ca2+ channel inhibitors that promote TNF/TNFR2 signaling.
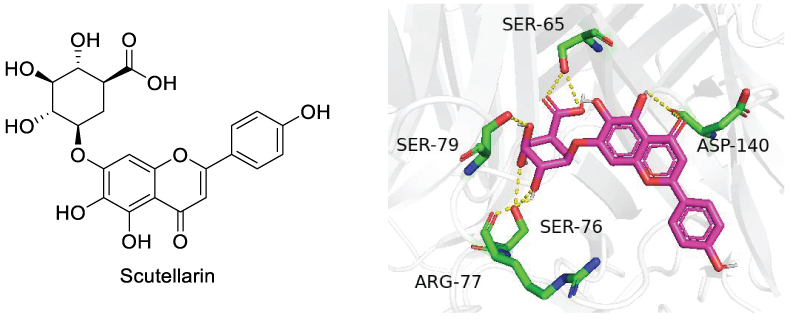
Scheme 15: Binding site of scutellarin with TNFR2.
Tetrandrine and scutellarin are good examples showing that natural products might be an important source for TNFR2 modulator discovery. Chen et al. identified another natural product, tacrolimus (TAC), that exhibits a therapeutic effect on psoriasis by interfering with the TNFR2 signaling pathway [69] (Scheme 16). The authors found that TAC treatment inhibited imiquimod-induced psoriasis in wild-type (WT) and TNFR1 knockout (TNFR1 KO) mice, but not in TNFR2 knockout (TNFR2 KO) mice. This therapeutic effect was induced by the expansion of MDSCs in a TNFR2-dependent manner.
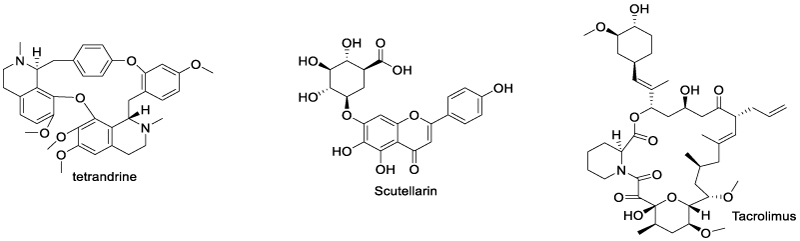
Scheme 16: TNF/TNFR2 transduction modulators sourced from natural products.
Another efficient strategy to mediate TNF-TNFR2 transduction is to focus on its downstream signaling pathway. In 2012, Wu et al. discovered that the MAPK signaling pathway was responsible for the TNF-TNFR2 stimulated expansion of Foxp3+ Tregs [70]. Some small molecule inhibitors of MAPK signal pathway, including SB203580 (P38 MAPK inhibitor), SP600125 (JNK inhibitor) and PD98059 (Erk1/2 inhibitor), potently suppressed TNF-TNFR2 induced replication of Tregs (Scheme 17).
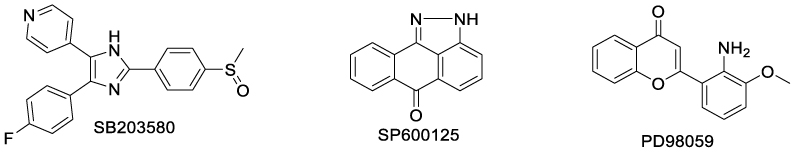
Scheme 17: MAPK pathway inhibitors that suppress TNF/TNFR2 induced replication of Tregs.
6. Emerging Trends and Future Perspectives
With growing knowledge of the pivotal roles that TNFRs play in pathological processes, TNFR modulators are emerging as the next-generation of treatment strategies against inflammation, immune regulation, and cancer. Although current TNFR modulators are dominated by macromolecular reagents such as antibodies, small-molecule TNFR modulators have emerged as a promising alternative due to their potential advantages of high selectivity, oral bioavailability, and tunable activity. In this brief review, we summarize small-molecule TNFR modulators reported in recent years. Based on pharmacological approaches, current strategies to develop small-molecule TNFR modulators include:
1. TNF ligands that prevent TNF from binding to TNFRs either by mimicking TNFR binding regions (C87) or by disrupting/disturbing TNF trimerization (SPD304 and SAR441566). This latter approach seems more promising as SAR441566 has been advanced to a clinic trials.
2. TNFR ligands binding to the key regions that interact with TNF, as with IV927 for TNFR1 and scutellarin for TNFR2.
3. Allosteric TNFRs modulators that regulate conformational changes by binding to non-active sites of TNFR and disabling its function, such as SGT11 and F002.
4. Antagonists that block receptor-ligand interactions or downstream signaling pathways (e.g. NF-κB, MAPK).
5. Protein-protein interaction (PPI) inhibitors that disrupt the assembly of TNFR complexes, such as SB200646.
6. TNF/TNFR-related upstream Ca2+ channel modulators, such as tetrandrine.
Compared with antibodies, small-molecule TNFR modulators offer advantages in cost, administration convenience, and tissue penetration. However, reports of small-molecule TNFR modulators are still rare, especially for TNFR2 modulators. Currently reported small-molecule TNFR modulators are generally less potent than antibodies, limiting their clinical application. Besides, challenges such as insufficient target selectivity, low in vivo efficacy, unsatifying stability and poor distribution/metabolism properties remain to be addressed. Future research may address these issues by integrating structural biology, computer-aided drug design, and optimization of drug-likeness properties to facilitate clinical translation. In the future, some emerging trends that might be applied to the small-molecule TNFR modulator development. Target protein degradation (TPD) such as PROTAC has been developed as a promising technology for inhibiting proteins that are difficult to target by traditional small-molecules [71]. This strategy might be applicable to discovery of challenging TNFR2 degrading agents. Two-pore channels (TPCs) has been proved to play a critical role in the modulation of expansion of TNFR2 expressing Tregs, and future research might focus on the development of (TPC) inhibitors or agonists. TNF-converting enzyme (TACE) modulators are also emerging as a novel way to balance the function between TNFR1 and TNFR2. With the aid of above-mentioned new techniques, it is expected that more small-molecule TNFR modulators might be advanced to clinic trials and eventually become an important alternative to antibodies in near future.
7. Funding
This work was funded by Guangdong S&T programme, 2023A0505030012; Guangzhou Key Research and Development Program, 202206010051.
8. References
- Loetscher H, Pan YC, Lahm HW, Gentz R, Brockhaus M, et al (1990). Molecular cloning and expression of the human 55 kd tumor necrosis factor receptor. Cell 61(2): 351-359.
- Wertz IE, Dixit VM (2009). Ubiquitin-mediated regulation of TNFR1 signaling. Cytokine Growth F R 19(3-4): 313-324.
- Fu Q, Shen Q, Tong J, Huang L, Cheng Y, et al (2021). Anti-tumor necrosis factor receptor 2 antibody combined with anti-PD-L1 therapy exerts robust antitumor effects in breast cancer. Front Cell Dev Biol 9: 720472.
- Ghods A, Mehdipour F, Shariat M, Talei AR, Ghaderi A (2021). Regulatory T cells express tumor necrosis factor receptor 2 with the highest intensity among CD4(+) T cells in the draining lymph nodes of breast cancer. Mol Immunol 137: 52-56.
- Mancusi A, Alvarez M, Piccinelli S, Velardi A, Pierini A (2019). TNFR2 signaling modulates immunity after allogeneic hematopoietic cell transplantation. Cytokine Growth Factor Rev 47: 54-61.
- Beldi G, Bahiraii S, Lezin C, Nouri Barkestani M, Abdelgawad ME, et al (2020). TNFR2 is a crucial hub controlling mesenchymal stem cell biological and functional properties. Front Cell Dev Biol 8: 596831.
- Madsen PM, Motti D, Karmally S, Szymkowski DE, Lambertsen KL, et al (2016). Oligodendroglial TNFR2 mediates membrane TNF-Dependent repair in experimental autoimmune encephalomyelitis by promoting oligodendrocyte differentiation and remyelination. J Neurosci 36(18): 5128-5143.
- Polz J, Remke A, Weber S, Schmidt D, Weber-Steffens D, et al (2014). Myeloid suppressor cells require membrane TNFR2 expression for suppressive activity. Immun Inflamm Dis 2(2): 121-130.
- Grell M, Wajant H, Zimmermann G, Scheurich P (1998). The type 1 receptor (CD120a) is the high-affinity receptor for soluble tumor necrosis factor. Proc Natl Acad Sci USA 95(2): 570-575.
- Grell M, Douni E, Wajant H, Lohden M, Clauss M, et al (1995). The transmembrane form of tumor necrosis factor is the prime activating ligand of the 80 kDa tumor necrosis factor receptor. Cell 83(5): 793-802.
- García-Domínguez M (2025). The Role of TNF-α in neuropathic pain: an immunotherapeutic perspective. Life 15(5): 785-807.
- Wajant H, Scheurich P (2011). TNFR1-induced activation of the classical NF-κB pathway. FEBS Journal 278(6): 862-876.
- Jang DI, Lee AH, Shin HY, Song HR, Park JH, et al (2021). The role of tumor necrosis factor alpha (TNF-α) in autoimmune disease and current TNF-α inhibitors in therapeutics. Int J Mol Sci 22(5): 2719-2735.
- Derksen V, Huizinga TWJ, van der Woude D (2017). The role of autoantibodies in the pathophysiology of rheumatoid arthritis. Semin Immunopathol 39(4):437-446.
- Britzen-Laurent N, Weidinger C, Stürzl M (2023). Contribution of blood vessel activation, remodeling and barrier function to Inflammatory Bowel diseases. Int J Mol Sci 24(6): 5517-5545.
- Zhou S, Ou R, Huang L, Moskophidis D (2002). Critical role for perforin-, Fas/FasL-, and TNFR1-mediated cytotoxic pathways in down-regulation of antigenspecific T cells during persistent viral infection. J Virol 76(2): 829-840.
- Alam MS, Gaida MM, Witzel HR, Otsuka S, Abbasi A, et al (2024). TNFR1 signaling promotes pancreatic tumor growth by limiting dendritic cell number and function. Cell Rep Med 5(9): 101696.
- Hayden MS, Ghosh S (2008). Shared principles in NF-kappaB signaling. Cell 132(3): 344-362.
- Scheidereit C (2006). Ikappa B kinase complexes: gateways to NF-kappaB activation and transcription. Oncogene 25(51): 6685-6705.
- Moatti A, Cohen JL (2021). The TNF-α/TNFR2 Pathway: Targeting a brake to release the anti-tumor immune response. Front Cell Dev Biol 9: 725473.
- Vanamee ES, Faustman DL (2017). TNFR2: a novel target for cancer immunotherapy. Trends Mol Med 23(11): 1037-1046.
- Yang Y, Islam MS, Hu Y, Chen X (2021). TNFR2: role in cancer immunology and immunotherapy. ImmunoTargets Ther 10: 103-122.
- Xu C, Ezzi SHA, Zou X, Dong Y, Alhaskawi A, et al (2025). The role of TNF in metabolic disorders and liver diseases. Cytokine 190: 156933.
- Watts TH, Yeung KKM, Yu T, Lee S, Eshraghisamani R (2025). TNF/TNFR superfamily members in costimulation of T cell responses—revisited. Annu Rev Immunol 43: 113-142.
- Williams RO, Clanchy FI, Huang YS, Tseng WY, Stone TW (2024). TNFR2 signalling in inflammatory diseases. Best Pract Res Cl Rh 38(2): 101941.
- Vanamee ES, Faustman DL (2017). TNFR2: A novel target for cancer immunotherapy. Trends Mol Med 23(11): 1037-1046.
- Chédotal H, Narayanan D, Povlsen K, Gotfredsen CH, Brambilla R, et al (2023). Small-molecule modulators of tumor necrosis factor signaling. Drug Discov Today 28(6): 103575.
- Davis JM, Colangelo J (2013). Small-molecule inhibitors of the interaction between TNF and TNFR. Future Med Chem 5(1): 69-79.
- Laura G, Rachele A, Marina C, Mariangela M, Alessandra R, et al (1992). Inhibitory effect of suramin on receptor binding and cytotoxic activity of tumor necrosis factor α. Int J Immunopharmacol 1992(14): 637-642.
- John J, Richard E, Alexander P, Tim G, Michael F, et al (2021). Highly accurate protein structure prediction with AlphaFold. Nature 596: 583-589.
- Dima K, Laurie EG, David RH, Tanggis B, Scott E M, et al (2015). The FTMap family of web servers for determining and characterizing ligand-binding hot spots of proteins. Nat Protoc 10: 733-755.
- Takasaki W, Kajino Y, Kajino K, Murali R, Greene MI (1997) Structure-based design and characterization of exocyclic peptidomimetics that inhibit TNF binding to its receptor. Nat Biotechnol 15: 1266-1270.
- Banner, DW, D’Arcy A, Janes W, Gentz R, Schoenfeld HJ, et al (1993). Crystal structure of the soluble human 55 kd TNF receptor-human TNF β complex: implications for TNF receptor activation. Cell 73: 431–445
- Saito H, Kojima T, Takahashi M, Horne WC, Baron R, et al (2007). A tumor necrosis factor receptor loop peptide mimic inhibits bone destruction to the same extent as anti-tumor necrosis factor monoclonal antibody in murine collagen-induced arthritis. Arthritis Rheum 56(4): 1164–1174
- Ma L, Gong H, Zhu H, Ji Q, Su P, et al (2014). A novel Small-molecule tumor necrosis factor inhibitor attenuates inflammation in a hepatitis mouse model. J Biol Chem 289(18): 12457-12466.
- He MM, Smith AS, Oslob JD, Flanagan WM, Braisted AC, et al (2005). Small-molecule inhibition of TNF-α. Science 310: 1022–1025
- Shen Q, Chen J, Wang Q, Deng X, Liu Y, et al (2014). Discovery of highly potent TNFα inhibitors using virtual screen. Eur J Med Chem 85:119-126.
- Yang Y, Liu Y, Sa K, Wang H, Yu L, et al (2023). Structure-based drug design and synthesis of pyrrolidine-2,5-diones as novel TNF-α inhibitors. J Chem Inf Model 63(12):3911-3924.
- O’Connell J, Porter J, Kroeplien B, Norman T, Rapecki S, et al (2019). Small molecules that inhibit TNF signalling by stabilising an asymmetric form of the trimer. Nature Commun 10:5795-5806.
- McMillan D, Martinez-Fleites C, Porter J, Fox D, Davis R, et al (2021). Structural insights into the disruption of TNF/TNFR1 signalling by small molecules stabilising a distorted TNF. Nature Commun 12:582-593.
- Vugler A, O’Connell J, Nguyen MA, Weitz D, Leeuw T, et al (2022). An orally available small molecule that targets soluble TNF to deliver anti-TNF biologic-like efficacy in rheumatoid arthritis. Front Pharmacol 13:1037983 .
- Xia K (2025). Recent advances in bridged structures as 3D bioisosteres of ortho-phenyl rings in medicinal chemistry applications. Chem Commun 61(35): 6417-6425.
- Dömling A, Holak TA (2025). Balinatunfib: A Clinical Oral Small Molecule TNFα Inhibitor. ChemMedChem 20: e202500258
- Nassr N, Rharbaoui F, Weitz D, Gassenhuber J, Rehberg M, et al (2025). First-in-Human single and multiple ascending dose studies of Balinatunfib, a small molecule inhibitor of TNFR1 signaling in healthy participants. Clin Pharmacol Ther 118(1):164-176.
- Carter PH, Scherle PA, Muckelbauer JK, Voss ME, Liu RQ, et al (2001). Photochemically enhanced binding of small molecules to the tumor necrosis factor receptor-1 inhibits the binding of TNF-α Proc Natl Acad Sci USA 98(21): 11879-11884
- Lo CH, Vunnam N, Lewis AK, Chiu TL, Brummel BE, et al (2017). An innovative high-throughput screening approach for discovery of small molecules that inhibit TNF receptors. SLAS Discovery 22(8):950-961.
- Gao M, Zhu H, Guo J, Lei Y, Sun W, Lin H (2022). Tannic acid through ROS/TNF-α/TNFR 1 antagonizes atrazine induced apoptosis, programmed necrosis and immune dysfunction of grass carp hepatocytes. Fish Shellfish Immun 131:312-322.
- Chen S, Feng Z, Wang Y, Ma S, Hu Z, et al (2017). Discovery of novel ligands for TNF‑α and TNF receptor‑1 through structure-based virtual screening and biological assay. J Chem Inf Model 57(5):1101−1111.
- Murali R, Cheng X, Berezov A, Du X, Schon A, et al (2005). Disabling TNF receptor signaling by inducedconformational perturbation of tryptophan-107. Proc Natl Acad Sci USA. 102:10970-10975.
- Rowe1 RK, Harrison JL, Zhang H, Bachstetter AD, Hesson DP, O’Hara BF, Greene MI, Lifshitz J (2018). Novel TNF receptor-1 inhibitors identified as potential therapeutic candidates for traumatic brain injury. J Neuroinflamm 15:154-167.
- Lo CH, Schaaf TM, Grant BD, Lim CKW, Bawaskar P, et al (2019). Noncompetitive inhibitors of TNFR1 probe conformational activation states. Sci Signal 12(592): eaav5637.
- Lo CH, Huber EC, Sachs JN (2020). Conformational states of TNFR1 as a molecular switch for receptor function. Protein Science 29(6): 1401-1415.
- Lo CH (2025). Targeting the inter-monomeric space of TNFR1 pre-ligand dimers: A novel binding pocket for allosteric modulators. Comput and Struct Biotec 27:1335-1341.
- Saddala MS, Huang H. Identifcation of novel inhibitors for TNFα, TNFR1 and TNFα-TNFR1 complex using pharmacophore-based approaches. J Transl Med 2019, 17: 215-230.
- Oli AN, Adejumo SA, Rowaiye AB, Ogidigo JO, Hampton-Marcell J, et al (2024). Tumour immunotherapy and applications of immunological products: a review of literature. J Immunol Res, 8481761.
- Tie Y, Tang F, Wei YQ, Wei XW. (2022) Immunosuppressive cells in cancer: mechanisms and potential therapeutic targets. J Hema Oncol 15(1): 61-93.
- Budhu S, Schaer DA, Li Y, Toledo-Crow R, Panageas K, et al (2017). Blockade of surface-bound TGF-β on regulatory T cells abrogates suppression of effector T cell function in the tumor microenvironment. Science Signaling 2017, 10(494): eaak9702.
- Xue Q, Peng W, Zhang S, Wei X, Ye L, et al (2024). Lactylation-driven TNFR2 expression in regulatory T cells promotes the progression of malignant pleural effusion. J ImmunoTher Cancer 12: e010040.
- Jin X, Zhang S, Wang N, Guan L, Shao C, et al (2022). High Expression of TGF-β1 contributes to hepatocellular carcinoma prognosis via regulating tumor immunity. Front Oncol 12: 861601.
- Huang L, Guo Y, Liu S, Wang H, Zhu J, et al (2021). Targeting regulatory T cells for immunotherapy in melanoma. Mol Biomed 2(1): 11-25.
- Marriott JB, Clarke IA, Dredge K, Muller G, Stirling D, et al (2002). Thalidomide and its analogues have distinct and opposing effects on TNF-α and TNFR2 during co-stimulation of both CD4+ and CD8+ T cells. Clin Exp Immunol 130: 75-84.
- Mukai Y, Nakamura T, Yoshikawa M, Yoshioka Y, Tsunoda Nakagawa S, et al (2010). Solution of the structure of the TNF-TNFR2 complex. Sci Signal 3: ra83.
- Shaikh F, He J, Bhadra P, Chen X, Siu SWI (2018). TNF receptor type II as an emerging drug target for the treatment of cancer, autoimmune diseases, and Graft-Versus-Host disease: current perspectives and in silico search for small molecule binders. Front Immunol 9: 1382.
- He T, Yang D, Li XQ, Jiang M, Islam MS, Chen S,et al (2020). Inhibition of two-pore channels in antigen-presenting cells promotes the expansion of TNFR2-expressing CD4+Foxp3+ regulatory T cells. Sci Adv 6(40): eaba6584.
- Liu QY, Karpinski E, Rao MR, Pang PKT (1991). Tetrandrine: A novel calcium channel antagonist inhibits type I calcium channels in neuroblastoma cells. Neuropharmacology 30(12A): 1325–1331.
- Sakurai Y, Kolokoltsov AA, Chen CC, Tidwell MW, Bauta WE, et al (2015). Two-pore channels control Ebola virus host cell entry and are drug targets for disease treatment. Science 347: 995-998.
- Chen S, Li R, Chen Y, Chou CK, Zhang Z (2022). Scutellarin enhances anti-tumor immune responses by reducing TNFR2-expressing CD4+Foxp3+ regulatory T cells. Biomed Pharmacother151:113187.
- Mei X, Ouyang H, Zhang H, Jia W, Lu B, et al (2023). Scutellarin suppresses the metastasis of triple-negative breast cancer via targeting TNFα/TNFR2-RUNX1-triggered G-CSF expression in endothelial cells. Biochem Pharmacol 217: 115808.
- Chen S, Liao P, Xi L, Yang Y, Wu W, et al (2022). The therapeutic effect of tacrolimus in a mousepsoriatic model is associated with the induction of myeloid-derived suppressor cells. Rheumatology Immunol Res 3(4): 190-197.
- Wu X, Chen Y, Han C, Gong Y, Bu D, et al (2012). Effects of Small-Molecule Inhibitors of TNFR2 Signaling Pathways On TNF-Mediated Expansion of CD4+FoxP3+ Regulatory T Cells. Blood 120(21): 4722.
- Gao H, Sun X, Rao Y (2020). PROTAC Technology: Opportunities and Challenges. ACS Med Chem Lett 11(3): 237−240.